Benchmarks¶
Algorithm Performance¶
To test the efficiency of ADAM’s implementations of the various algorithms that are used in our variant calling pipeline, we ran strong scaling experiments. In this experiment, we used the high coverage genome NA12878 from the 1000 Genomes project. We held the executor configuration constant, and stepped from one to four executors before doubling until we reached 32 executors. Each executor had 16 hyperthreaded cores (32 concurrent threads) and was allocated 200 gigabytes of memory.
In general, most of the algorithms in ADAM scale linearly as the amount of compute resources is increased. The significant exception is BQSR, which is explained by the large broadcast which runs during BQSR. The broadcast introduces a serial step that runs for several minutes, as the multiple gigabyte mask table is broadcast to all of the nodes in the cluster. The figures below break these results down and compares them to the latest implementation of these algorithms in the GATK4.
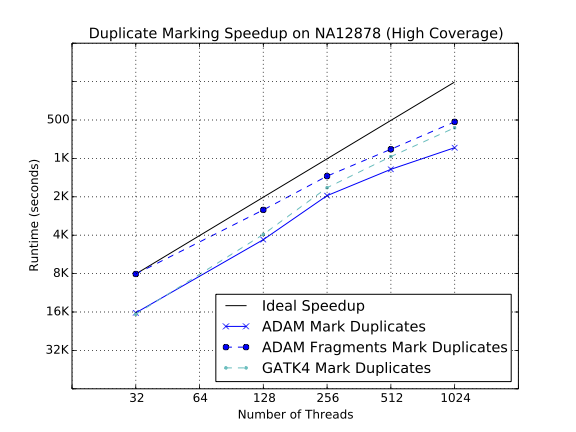
Strong scaling characteristics of duplicate marking
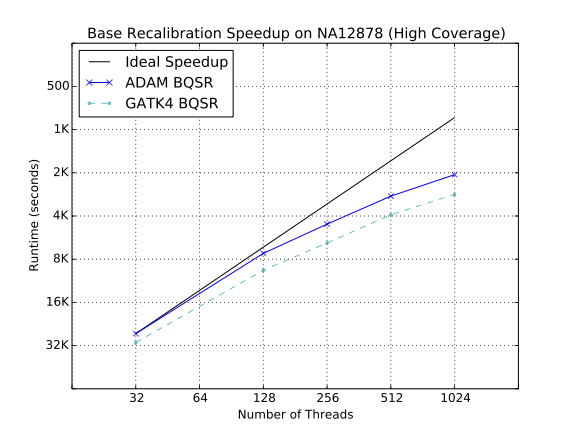
Strong scaling characteristics of base quality recalibration
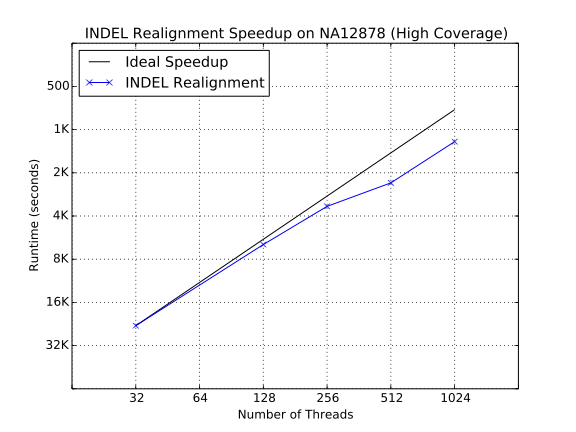
Strong scaling characteristics of INDEL Realignment
As our benchmarks demonstrate, ADAM’s implementations of the various algorithms in the standard germline reads-to-variants pipeline are more performant than the same algorithms implemented in the GATK4. ADAM outperforms the GATK when running equivalent implementations of BQSR (1.4x speedup) and duplicate marking (1.1x speedup). To improve the performance of duplicate marking, we added a variant of the duplicate marking algorithm that operates on reads that are grouped by sequencing fragment. By optimizing for reads that are grouped with the other reads from their fragment, we can eliminate the first shuffle in duplicate marking, which reassembles reads by read-pair. This optimization is common in duplicate markers, as paired-end aligners will typically emit a SAM or BAM file where consecutive reads are from the same sequencing fragment.